O'Reilly PG, Claffey NM. A history of oral sepsis as a cause of disease. Periodontol 2000. 2000; 23:13-18
Mayo CH. Focal infection of dental origin. Dental Cosmos. 1922; 64:1206-1208
Hunter W. Oral sepsis as a cause of disease. Br Med J. 1900; 2:215-216
Mattila KJ, Nieminen MS, Valtonen VV Association between dental health and acute myocardial infarction. Br Med J. 1989; 298:779-781
Scannapieco FA. Position paper of The American Academy of Periodontology: periodontal disease as a potential risk factor for systemic diseases. J Periodontol. 1998; 69:841-850
Mustapha IZ, Debrey S, Oladubu M, Ugarte R. Markers of systemic bacterial exposure in periodontal disease and cardiovascular disease risk: a systematic review and meta-analysis. J Periodontol. 2007; 78:2289-2302
Heo SM, Haase EM, Lesse AJ, Gill SR, Scannapieco FA. Genetic relationships between respiratory pathogens isolated from dental plaque and bronchoalveolar lavage fluid from patients in the intensive care unit undergoing mechanical ventilation. Clin Infect Dis: an official publication of the Infectious Diseases Society of America. 2008; 47:1562-1570
Han YW. Oral health and adverse pregnancy outcomes – what's next?. J Dent Res. 2011; 90:289-293
Molina CA, Ojeda LF, Jiménez MS Diabetes and periodontal diseases: an established two-way relationship. J Diabetes Mellitus. 2016; 6:209-229
Kaur S, Bright R, Proudman SM, Bartold PM. Does periodontal treatment influence clinical and biochemical measures for rheumatoid arthritis? A systematic review and meta-analysis. Semin Arthritis Rheum. 2014; 44:113-122
Strauss J, Kaplan GG, Beck PL Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011; 17:1971-1978
Castellarin M, Warren RL, Freeman JD Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012; 22:299-306
Han P, Sun D, Yang J. Interaction between periodontitis and liver diseases. Biomed Rep. 2016; 5:267-276
Beck J, Garcia R, Heiss G, Vokonas PS, Offenbacher S. Periodontal disease and cardiovascular disease. J Periodontol. 1996; 67:1123-1137
Thoden van Velzen SK, Abraham-Inpijn L, Moorer WR. Plaque and systemic disease: a reappraisal of the focal infection concept. J Clin Periodontol. 1984; 11:209-220
Roberts GJ. Dentists are innocent! “Everyday” bacteremia is the real culprit: a review and assessment of the evidence that dental surgical procedures are a principal cause of bacterial endocarditis in children. Pediatr Cardiol. 1999; 20:317-325
Que YA, Moreillon P. Infective endocarditis. Nat Rev Cardiol. 2011; 8:322-336
Douglas CWI, Heath J, Hampton KK, Preston FE. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol. 1993; 39:179-182
Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. J Periodontol. 2000; 71:1554-1560
Li X, Kolltveit KM, Tronstad L, Olsen I. Systemic diseases caused by oral infection. Clin Microbiol Rev. 2000; 13:547-558
Van Dyke TE, Dowell VR, Offenbacher S, Snyder W, Hersh T. Potential role of microorganisms isolated from periodontal lesions in the pathogenesis of inflammatory bowel disease. Infect Immun. 1986; 53:671-677
Bhatnagar P, Wickramasinghe K, Williams J, Rayner M, Townsend N. The epidemiology of cardiovascular disease in the UK 2014. Heart. 2015; 101:1182-1189
Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999; 340:115-126
Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011; 473:317-325
Scannapieco FA, Bush RB, Paju S. Associations between periodontal disease and risk for atherosclerosis, cardiovascular disease, and stroke. A systematic review. Ann Periodontol/The American Academy of Periodontology. 2003; 8:38-53
Burazor I, Vojdani A. Chronic exposure to oral pathogens and autoimmune reactivity in acute coronary atherothrombosis. Autoimmune Dis. 2014; 2014
Syrjanen J, Peltola J, Valtonen V, Iivanainen M, Kaste M, Huttunen JK. Dental infections in association with cerebral infarction in young and middle-aged men. J Intern Med. 1989; 225:179-184
Hashemipour MA, Afshar AJ, Borna R, Seddighi B, Motamedi A. Gingivitis and periodontitis as a risk factor for stroke: a case-control study in the Iranian population. Dental Res J (Isfahan). 2013; 10:613-619
Rendu F, Brohard-Bohn B. The platelet release reaction: granules' constituents, secretion and functions. Platelets. 2001; 12:261-273
Bennett JS, Berger BW, Billings PC. The structure and function of platelet integrins. J Thromb Haemost. 2009; 7:200-205
Kerrigan SW. The expanding field of platelet-bacterial interconnections. Platelets. 2015; 26:293-301
Wu T, Yeaman MR, Bayer AS. In vitro resistance to platelet microbicidal protein correlates with endocarditis source among bacteremic staphylococcal and streptococcal isolates. Antimicrob Agents Chemother. 1994; 38:729-732
Deng L, Bensing BA, Thamadilok S Oral streptococci utilize a siglec-like domain of serine-rich repeat adhesins to preferentially target platelet sialoglycans in human blood. PLoS Pathogens. 2014; 10
Svensson L, Baumgarten M, Mörgelin M, Shannon O. Platelet activation by Streptococcus pyogenes leads to entrapment in platelet aggregates, from which bacteria subsequently escape. Infect Immun. 2014; 82:4307-4314
Clawson CC. Platelet interaction with bacteria. 3. Ultrastructure. Am J Pathol. 1973; 70:449-471
Clawson CC, Rao GH, White JG. Platelet interaction with bacteria. IV. Stimulation of the release reaction. Am J Pathol. 1975; 81:411-420
Clawson CC, White JG. Platelet interaction with bacteria. II. Fate of the bacteria. Am J Pathol. 1971; 65:381-397
Clawson CC, White JG. Platelet interaction with bacteria. I. Reaction phases and effects of inhibitors. Am J Pathol. 1971; 65:367-380
Kerrigan S, Cox D. Platelet-bacterial interactions as therapeutic targets in infective endocarditis. In: Breijo-Marquez FR (ed). : Intech Publishers; 2012
Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol. 2005; 129:101-109
Kerrigan SW, Clarke N, Loughman A, Meade G, Foster TJ, Cox D. Molecular basis for Staphylococcus aureus-mediated platelet aggregate formation under arterial shear in vitro. Arterioscler Thromb Vasc Biol. 2008; 28:335-340
Siegel I, Cohen S. Action of staphylococcal toxin on human platelets. J Infect Dis. 1964; 114:488-502
Schubert S, Schwertz H, Weyrich AS Staphylococcus aureus α-toxin triggers the synthesis of B-cell lymphoma 3 by human platelets. Toxins. 2011; 3:120-133
Jakubovics NS, Kerrigan SW, Nobbs AH Functions of cell surface-anchored antigen I/II family and Hsa polypeptides in interactions of Streptococcus gordonii with host receptors. Infect Immun. 2005; 73:6629-6638
Kerrigan SW, Jakubovics NS, Keane C Role of Streptococcus gordonii surface proteins SspA/SspB and Hsa in platelet function. Infect Immun. 2007; 75:5740-5747
Xiong YQ, Bensing BA, Bayer AS, Chambers HF, Sullam PM. Role of the serine-rich surface glycoprotein GspB of Streptococcus gordonii in the pathogenesis of infective endocarditis. Microb Pathog. 2008; 45:297-301
Takahashi Y, Konishi K, Cisar JO, Yoshikawa M. Identification and characterization of hsa, the gene encoding the sialic acid-binding adhesin of Streptococcus gordonii DL1. Infect Immun. 2002; 70:1209-1218
Keane C, Petersen H, Reynolds K Mechanism of outside-in αIIbβ3-mediated activation of human platelets by the colonizing bacterium, Streptococcus gordonii. Arterioscler Thromb Vasc Biol. 2010; 30:2408-2415
Petersen HJ, Keane C, Jenkinson HF Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infect Immun. 2010; 78:413-422
Keane C, Petersen HJ, Tilley DO Multiple sites on Streptococcus gordonii surface protein PadA bind to platelet GPIIbIIIa. J Thromb Haemost. 2013; 110:1278-1287
Haworth J, Jenkinson HF, Petersen H Concerted functions of Streptococcus gordonii surface protein PadA and Hsa mediate activation of human platelets and interactions with extracellular matrix. Cell Microbiol. 2016; https://doi.org/10.1111/cmi.12667
Mancini S, Menzi C, Oechslin F, Moreillon P, Entenza JM. Antibodies targeting Hsa and PadA prevent platelet aggregation and protect rats against experimental endocarditis induced by Streptococcus gordonii. Infect Immun. 2016; 84:3557-3563
Prophylaxis against infective endocarditis: antimicrobial prophylaxis against infective endocarditis in adults and children undergoing interventional procedures. NICE Clinical Guideline No 64. 2008. https://www.nice.org.uk/guidance/cg64/chapter/recommendations (Accessed November 2016)
Thornhill MH, Dayer MJ, Jones S, Prendergast B, Baddour LM, Lockhart PB. The effect of antibiotic prophylaxis guidelines on incidence of infective endocarditis. Can J Cardiol. 2016; 32
Thornhill MH, Dayer M, Lockhart PB Prophylaxis guidelines: Plea to NICE. Br Dent J. 2016; 221:2-3
Alderson P, Baker M. Prophylaxis guidelines: repeated points. Br Dent J. 2016; 220
Alderson P, Baker M. Prophylaxis guidelines: repeated points. Br Dent J. 2016; 220
Chambers JB, Thornhill M, Shanson D, Prendergast B. Antibiotic prophylaxis of endocarditis: a NICE mess. Lancet Infect Dis. 2016; 16:275-276
Chambers J, Thornhill M, Shanson D, Prendergast B. Antibiotic prophylaxis of endocarditis – Authors' reply. Lancet Infect Dis. 2016; 16:774-775
Million M, Grisoli D, Griffiths K, Raoult D. Antibiotic prophylaxis of endocarditis. Lancet Infect Dis. 2016; 16:773-774
Main BG, Adair SR. The changing face of informed consent. Br Dent J. 2015; 219:325-327
Thornhill MH, Dayer M, Lockhart PB A change in the NICE guidelines on antibiotic prophylaxis. Br Dent J. 2016; 221:112-114
Song M, O'Donnell JA, Bekhuis T, Spallek H. Are dentists interested in the oral-systemic disease connection? A qualitative study of an online community of 450 practitioners. BMC Oral Health. 2013; 13
Friedewald VE, Kornman KS, Beck JD The American Journal of Cardiology and Journal of Periodontology Editors' Consensus: periodontitis and atherosclerotic cardiovascular disease. Am J Cardiol. 2009; 104:59-68
Oral hygiene as a risk factor in infective endocarditis Jennifer A Haworth Richard G Mears Howard F Jenkinson Steve W Kerrigan Angela H Nobbs Dental Update 2024 44:9, 707-709.
Authors
Jennifer AHaworth
PhD
Academic Clinical Lecturer, Bristol Dental School, University of Bristol, Lower Maudlin Street, BS1 2LY, Bristol, UK
There are many known associations between oral and systemic diseases. This review paper summarizes the proposed mechanisms underlying the links between dental disease and cardiovascular disease before introducing recent research regarding bacteria-platelet interactions. New protein factors have been identified on dental plaque bacteria. One of these, PadA, triggers blood to clot. This research provides new information about how Streptococcus bacteria and platelets interact and could lead to the development of new ways to control the formation of blood clots caused by micro-organisms that access the bloodstream.
CPD/Clinical Relevance: This article aims to provide the whole dental team with an overview of bacteria-platelet interactions. This is of particular relevance to infective endocarditis and the recent change in wording to the NICE antibiotic prophylaxis guidelines in the UK.
Article
The mouth is often referred to as a window into the health of the body, but the idea of oral diseases being a risk factor for systemic diseases is not a new concept. The importance of oral hygiene was noted by the Ancient Greeks1 and Hippocrates recorded two cases where eradication of mouth infections relieved patients of rheumatic joint problems.2 The British surgeon, William Hunter, is regarded as one of the first in the 20th century to emphasize the role of oral sepsis in leading to generalized diseases3 and removal of infected teeth was common. In contrast, the second half of the 20th century saw increased conservation and restoration of teeth, with relatively little published about possible links between oral and systemic diseases. However, a seminal study in 1989 reported that dental health was significantly worse in patients with acute myocardial infarction, after adjusting for risk factors.4 Subsequent papers re-ignited the debate and the turn of the 21st century saw a renewed scientific interest in the links between oral and systemic diseases, with increasing awareness that the mouth can act as a reservoir for pathogenic bacteria and their products.
Of particular interest are the oral associations with cardiovascular disease, the most common cause of death worldwide. Classical risk factors (hypertension, hypercholesterolaemia, diabetes, ethnicity, smoking, family history, increased body weight, alcohol, lack of physical activity) only account for up to two-thirds of cardiovascular disease cases5 and evidence is emerging that chronic oral infection and inflammation may be other risk factors.6
The aim of this paper is to:
Provide an overview of the proposed mechanisms linking oral and systemic diseases;
Summarize the known associations between oral bacteria and heart disease;
Describe recent research into interactions between oral bacteria and platelets, particularly relevant to infective endocarditis.
Mechanisms linking oral and systemic diseases
The reported associations between oral and systemic conditions are numerous and include the following diseases: heart disease,6 ventilator-associated pneumonia,7 adverse pregnancy outcome,8 diabetes mellitus,9 rheumatoid arthritis,10 inflammatory bowel disease,11 colorectal cancer12 and liver diseases.13
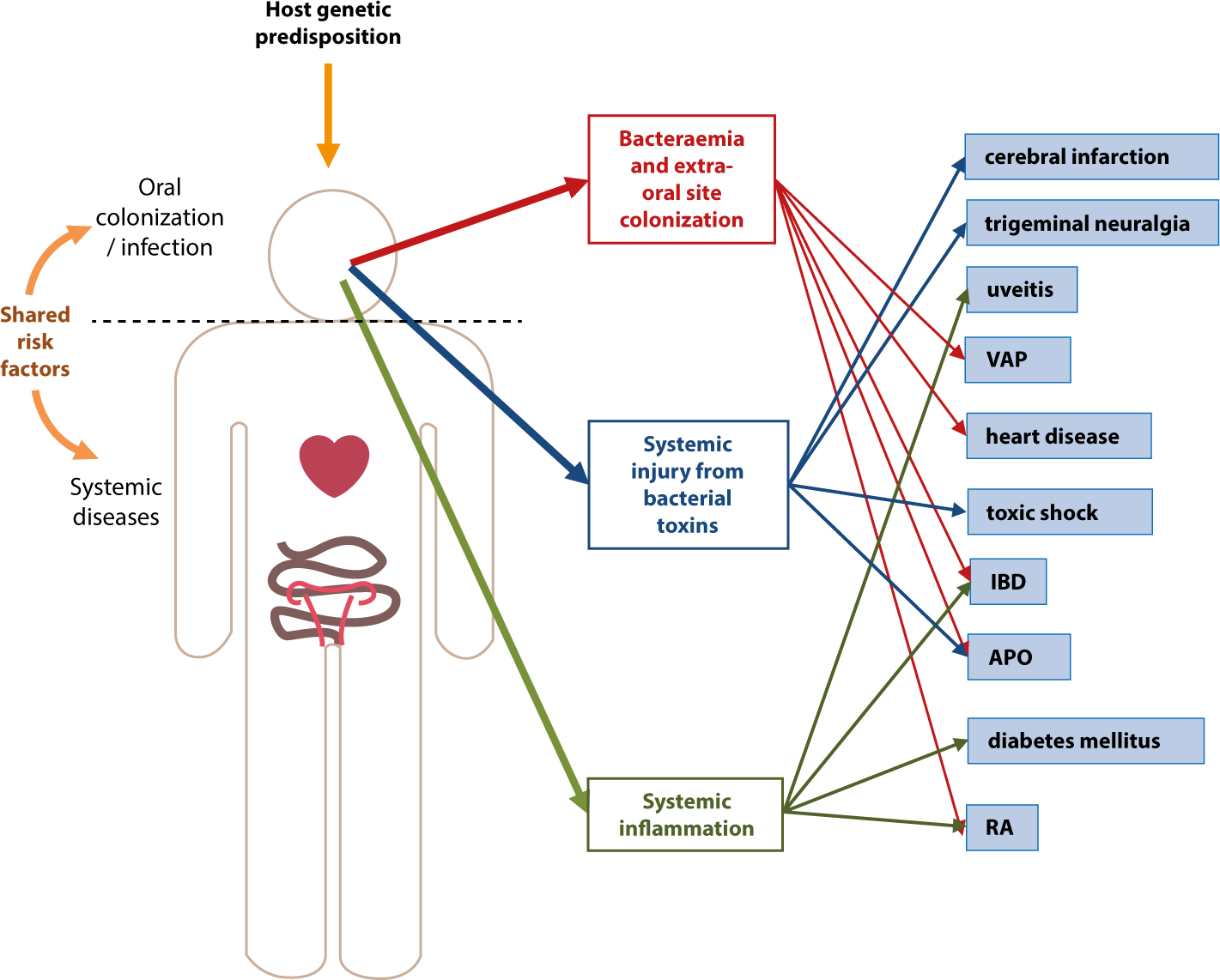
Shared risk factors for oral disease and systemic diseases include age, male gender, alcohol consumption, smoking, hypertension, social isolation, stress and diabetes.4,14 It is generally accepted that associations between oral and systemic diseases exist, but causation is more difficult to determine. In addition to shared risk factors, three possible mechanisms for the link between oral and systemic diseases have been proposed,15 described below and in Figure 1.
Figure 1. Diagrammatic representation of the mechanisms linking oral and systemic diseases. VAP = ventilator-associated pneumonia; IBD = inflammatory bowel disease; APO = adverse pregnancy outcome; RA = rheumatoid arthritis.
Bacteraemia and extra-oral site colonization
Entry of oral bacteria into the bloodstream (bacteraemia) during normal physiological activities may be as important in the pathogenesis of infective endocarditis (IE) as dentistry-induced bacteraemia.16 Cumulative levels of bacteraemia have been calculated to be approximately 100,000 times higher during physiological activities, such as chewing or flossing, than after a single dental extraction.17 Oral streptococci, commonly implicated in IE, are often isolated from blood culture18 and periodontal pathogens have been found in atherosclerotic plaques.19
Systemic injury as a result of bacterial toxins
The production of toxins by oral bacteria can cause direct damage to the host. One well-known example of an endotoxin is lipopolysaccharide (LPS), found in the outer membrane of Gram-negative bacteria. LPS can contribute to the induction of toxic shock, fever and apoptosis.20
Systemic inflammation caused by the immunological response to oral micro-organisms
Immune complexes form when soluble antigens react with circulating antibodies. The deposition of these immune complexes leads to chronic or acute inflammation, with raised levels of inflammatory mediators such as tumour necrosis factor-α, interleukin-1β and the vasodilator prostaglandin E2.21 Another acute-phase protein that activates the complement system during inflammation and is often used as a clinical marker of systemic inflammation is C-reactive protein (CRP).20 Several conditions have been associated with systemic inflammation caused by oral micro-organisms, for example inflammatory bowel disease, uveitis and Behçet's syndrome.21, 22
Cardiovascular disease
Cardiovascular disease is a term used to describe a spectrum of diseases of the circulatory system including myocardial infarction and stroke. Despite increased promotion of healthy lifestyles and widespread use of anti-cholesterol medication, cardiovascular disease continues to be a major burden in the UK.23
Atherosclerosis is characterized by artery wall thickening as a result of accumulation of fatty material, but there is also a major inflammatory component.24
The development of an atherosclerotic lesion is shown in Figure 2. Periodontal disease may have a moderate association with atherosclerosis, and oral micro-organisms have been detected in atherosclerotic lesions.19,26
Figure 2. The development of an atherosclerotic lesion (adapted from Libby et al, 2011).25(a) The normal artery. Three layers are found: the tunica intima, which is lined by endothelial cells; the tunica media, which comprises smooth muscle cells in an extracellular matrix; and the tunica adventitia, which contains mast cells and fibroblasts. (b) Initial atherosclerotic changes. Activated endothelial cells capture monocytes and these migrate into the tunica intima. They mature to become macrophages and when they take up lipid they become known as foam cells. (c) Lesion progression. Smooth muscle cells migrate to the tunica intima and collagen and other extracellular matrix molecules are produced. Cholesterol crystals and microvessels form and macrophages and smooth muscle cells undergo apoptosis. (d) Disruption of atherosclerotic plaque and thrombosis. The lipid core is an accumulation of extracellular lipid from dead cells. The fibrous cap of the plaque ruptures and formation of a thrombus is triggered.
Myocardial infarction
Acute myocardial infarction (MI) is a clinical manifestation of atherosclerosis. Serum antibody levels to oral anaerobic bacteria have been found to be higher in MI subjects compared to a control group. Increased CRP levels were also found in the MI group.27 This adds to the body of evidence that inflammation caused by oral bacteria plays a significant role in atherosclerosis and MI.
Stroke
Stroke is the loss of brain function due to a disturbance in brain blood supply, caused by thrombotic ischaemia or haemorrhage. Stroke is a recognized complication of IE28 and associations have been found between oral infections and stroke.29,30
Disseminated intravascular coagulation and sepsis
Disseminated intravascular coagulation (DIC) is a life-threatening condition that occurs in response to tissue damage or infection. It results in fibrin deposition and obstruction in the microvasculature, causing organ dysfunction.31 It has been proposed that bacteraemia secondary to periodontal disease potentially contributes to the occurrence of a DIC-like disease presentation in the immunocompromised.32 Sepsis is a systemic inflammatory response to infection and occurs when a controlled initial response to an infection becomes exaggerated and deregulated.33 Sepsis may escalate to septic shock if untreated. Viridans group streptococci entering the circulation from the mouth can cause septic shock in immunosuppressed patients.34
Infective endocarditis (IE)
Infective endocarditis is characterized by the formation on heart valves of infected masses or vegetations containing bacteria, fibrin and platelets (Figure 3). There are over 2000 cases of IE in the UK annually and the incidence is rising.35 Infective endocarditis has a subtle clinical presentation, usually including fever, malaise and weight loss.35 The modified Duke criteria, including blood cultures and echocardiography, are used in diagnosis.17 The treatment of IE patients is co-ordinated by a multidisciplinary team and aims to eradicate the implicated micro-organisms by antimicrobial drug treatment alone, or in conjunction with surgery. The use of antimicrobials poses several problems, such as poor penetration into vegetations, resulting in the need for high serum concentrations of antimicrobial agents.36 Bacterial resistance to antibiotics is also increasing, making IE a difficult condition to treat successfully. Mortality is high with 15–20% of IE patients dying during their initial hospital admission.35 Of those who survive, 10–15% die during the following year.35
Figure 3. Diagrammatic representation of the heart. The four heart valves are highlighted and infected vegetations are shown at the mitral valve and at a non-valve endocardial site. The direction of blood flow is denoted by arrows. The heart valves control the flow of the blood through the heart chambers. Pressure differences generated within the heart, along with muscle fibres, control the opening and closing of the valves. Oxygen-poor blood from the body enters the heart from the inferior and superior vena cava vessels into the right atrium. Blood flows into the right ventricle from the right atrium through the tricuspid valve, which then closes when the ventricle is full. When the right ventricle contracts, blood leaves the heart through the pulmonary valve entering the pulmonary arteries, where it continues to the lungs for oxygenation. Oxygen-rich blood returns to the left atrium from the pulmonary veins. Blood enters the left ventricle through the mitral valve. When the left ventricle contracts blood enters the aorta through the aortic valve. IE vegetations can be found at each of the valve sites highlighted or at multiple valve sites simultaneously. Both native and prosthetic valves can be affected.
Endocarditis can be classified according to the site (right- or left-sided), type of valve involved (native or prosthetic), or timing (early or late). Left-sided native valve endocarditis is the most common form of IE and occurs in people with pre-existing lesions of the heart, such as congenital heart defects or rheumatic fever-induced damage.
Streptococci, enterococci and Staphylococcus aureus as a group are responsible for more than 80% of cases of IE37 and oral viridans group streptococci are causative agents in 35–45% of cases of IE.38 Oral streptococci are the predominant IE pathogens in the general population, whereas S. aureus is more often found in healthcare-associated IE.17 New ‘at risk’ groups have emerged in recent years, such as IV drug users, patients with valve sclerosis and haemodialysis patients.37
Platelets
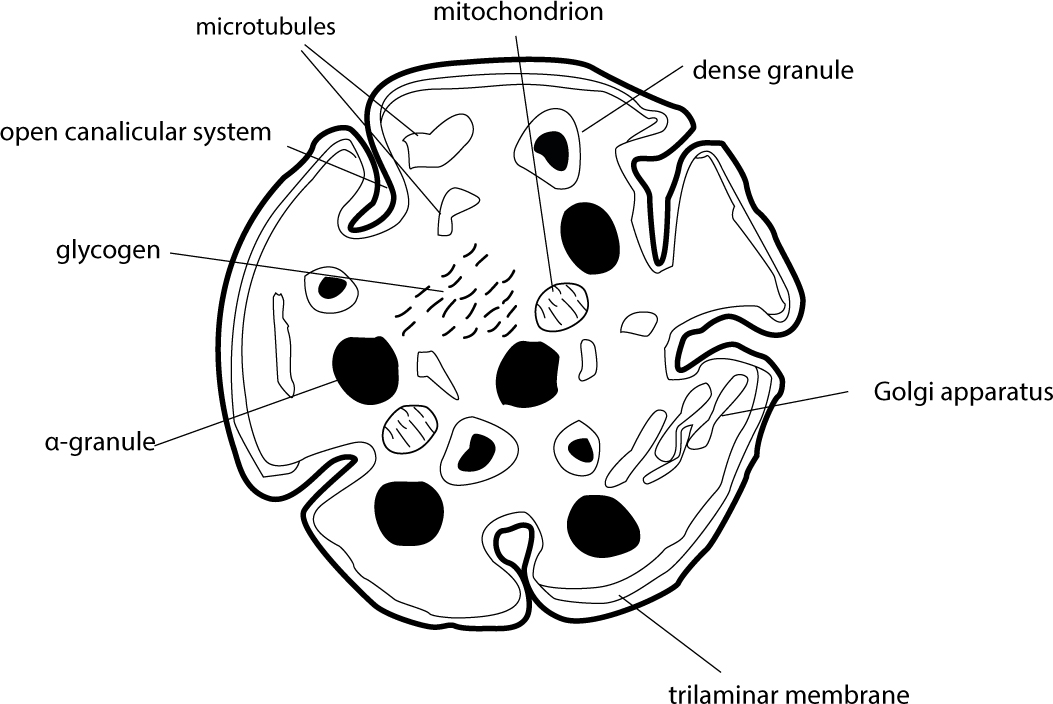
Platelets are anucleate cell fragments, formed from their precursor megakaryocytes in the bone marrow (Figure 4). They are approximately 2–3 µm in diameter and have a lifespan of 5–9 days.39 The platelet cytoplasm contains the endoplasmic reticulum, Golgi apparatus, mitochondria, granules and contractile proteins.40 The granules are stores of important clotting mediators such as adenosine diphosphate, serotonin, Ca2+ (dense granules), fibrinogen, vitronectin and von Willebrand factor (α-granules).41 A large number of platelet membrane receptors are necessary for haemostasis. Of particular note are the integrins, a family of heterodimers that mediate cell-cell or cell-substratum interactions by linking extracellular signals to intracellular pathways.42 Platelets typically circulate in a resting state at the periphery of the blood vessel, ready to respond to vessel wall damage. When they encounter exposed subendothelial tissues at the site of vessel wall damage, they adhere, become activated, aggregate and recruit other platelets to the site to form the platelet clot. Being the first cells present at injury sites also means that platelets are ideally placed to recruit immune cells. Evidence is emerging of the critical importance of platelets in orchestrating the immune response.43
Figure 4. The ultrastructure of a platelet. Platelets are anucleate cell fragments, measuring 2–3 µm in diameter. The trilaminar membrane maintains the integrity of the cytoplasm and invaginations of the membrane form the canalicular system. Dense granules contain adenosine diphosphate, serotonin and Ca2+ ions, and α-granules contain fibrinogen, vitronectin and von Willebrand factor.
Bacteria-platelet interactions
The cardiovascular system is normally a sterile environment in health. When bacteria enter the circulation, platelets can direct the immune response to manage exposure to bacterial pathogens. Bacteria-platelet binding triggers platelet activation and the secretion of antimicrobial peptides from platelets, although some bacteria have become resistant to these peptides.44 The immune platelet response in itself can become part of the pathogenic process leading to the formation of septic thrombi. Bacteria-induced platelet activation and aggregation allows bacteria to survive in infected thrombi, effectively hidden from the rest of the immune system by the platelet clot. This has been described as a ‘Trojan Horse’ effect for streptococcal-platelet interactions.45 Some bacteria are able to mediate the formation of unstable platelet aggregates from which microbial cells subsequently escape, allowing them to evade host immune responses and disseminate through the circulation during invasive infection.46
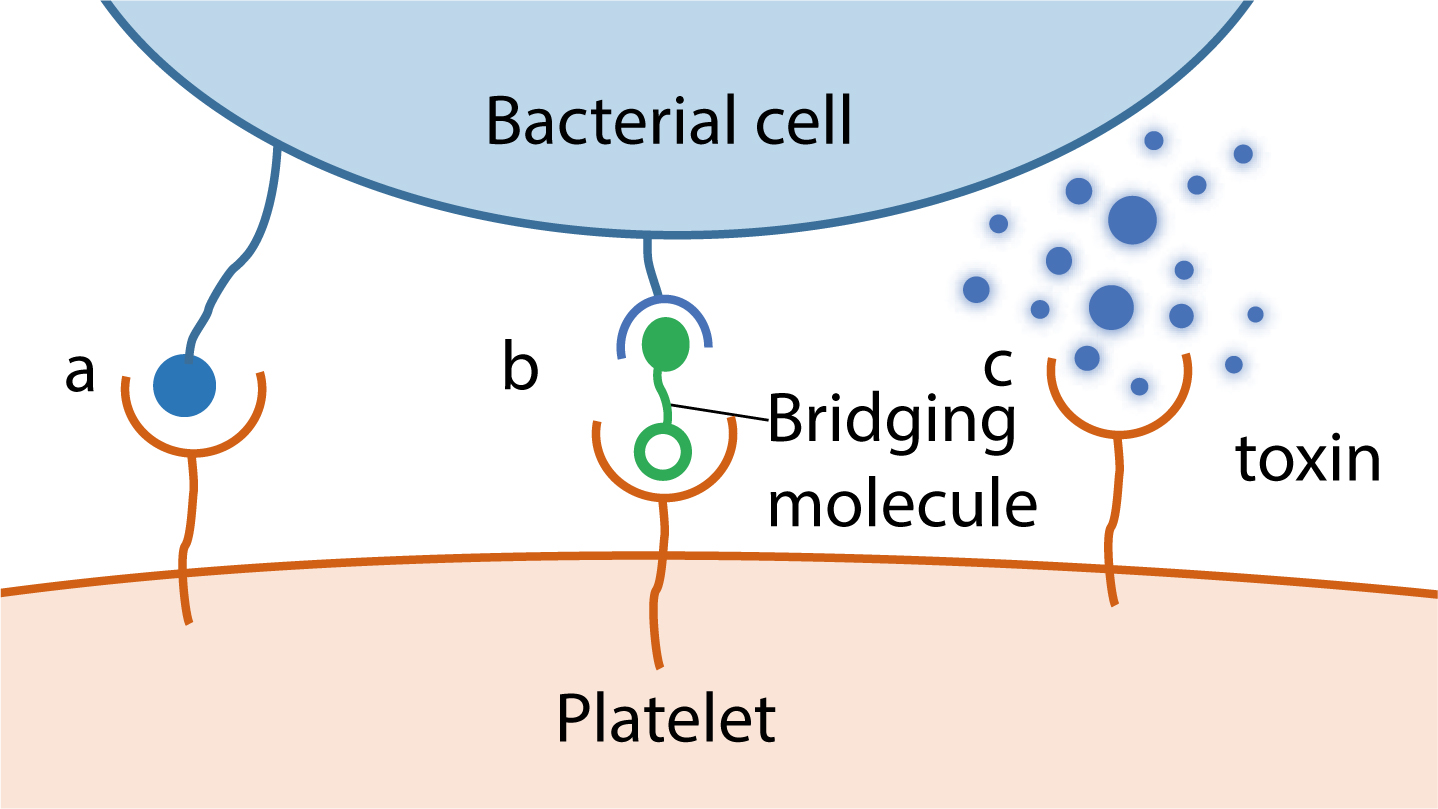
The study of bacteria-platelet interactions only gained momentum in the last 40 years following the publication of a series of papers by Clawson et al.47,48,50 Bacteria can interact with platelets via multiple mechanisms and one micro-organism may produce several factors capable of interacting with platelets simultaneously. This makes it difficult to study the role of individual receptors. However, three general modes of interaction between bacteria and platelets (Figure 5) have been described.51
Figure 5. Summary of the interactions between bacteria and platelets: (a) Direct interaction; (b) Indirect interaction and (c) Bacterial toxin-mediated interaction. Streptococcus gordonii employs the direct inter-action (a); Streptococcus pyogenes utilizes the indirect bridging mechanism (b); Staphylococcus aureus employs all three mechanisms (a, b, c).
Direct binding of platelet receptor to bacterial surface component
The direct interaction of a specific bacterial protein with target platelet receptors leads to platelet adhesion or activation. Streptococcus serine-rich repeat proteins bind directly to platelet glycoprotein Ib and mediate platelet rolling and adhesion under flow conditions.52
Indirect binding of platelet receptor to bacterium via a bridging plasma protein, such as fibrinogen or complement
An indirect interaction with platelets is shown by S. aureus Clumping factor A (ClfA). Simultaneous binding of ClfA to fibrinogen and antibody, which interacts with integrin αIIbβ3 and FcγRIIa on the platelet surface, is essential for platelet aggregate formation.53
Secretion of bacterial toxins that interact with platelets
Staphylococcus aureus releases a toxin known as α-toxin that causes platelet aggregation and shape change.54 This toxin has more recently been shown to induce production of B-cell lymphoma 3 in platelets, a factor thought to be important in the development of IE.55
Streptococcus gordonii-platelet interactions
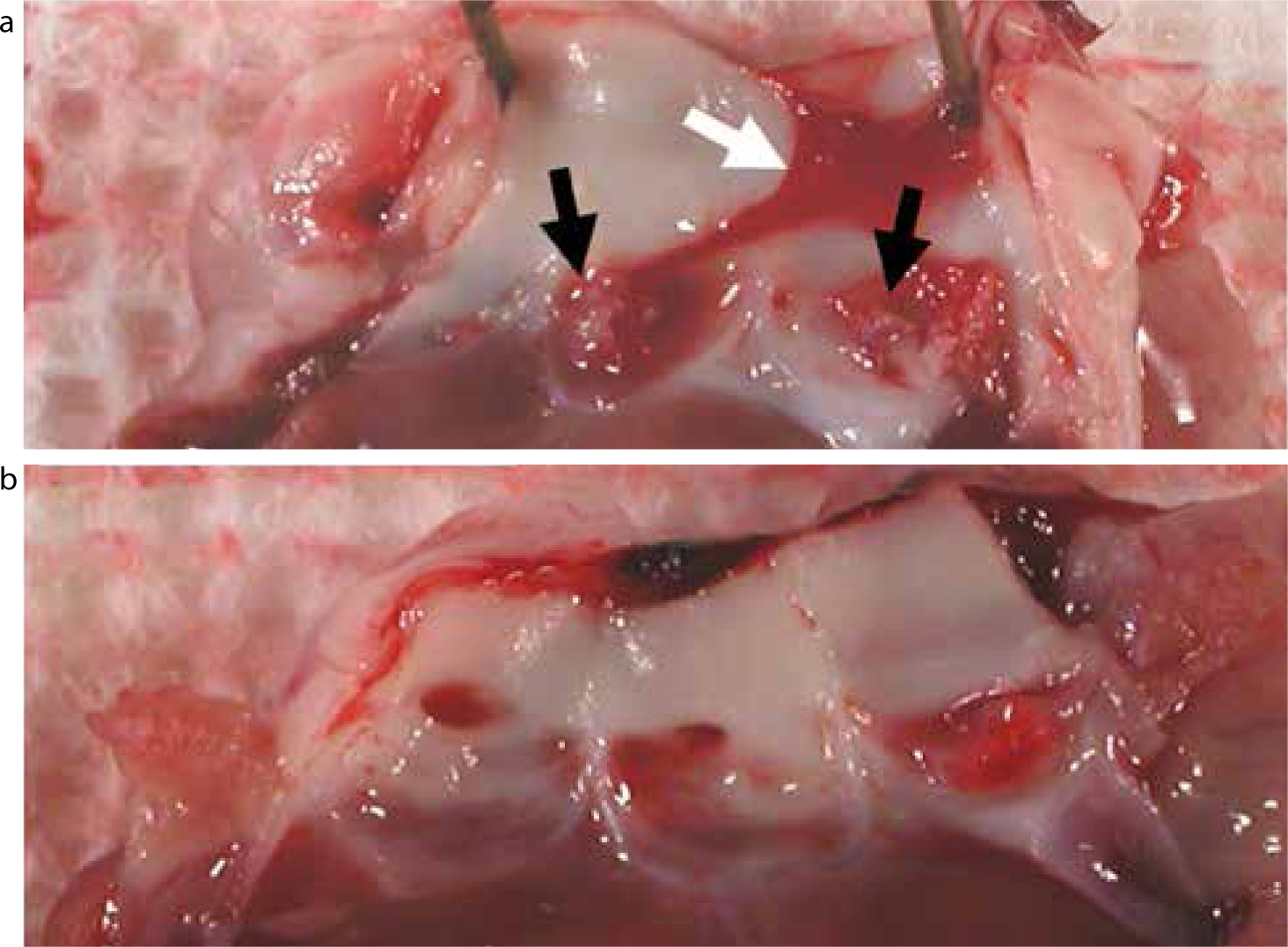
Oral streptococci are major mediators of IE, and have numerous surface proteins capable of modulating attachment to surfaces within the oral cavity, endothelial tissue, extracellular matrix and platelets.56 The interactions of platelets with S. gordonii, a ubiquitous oral bacterium often implicated in IE,18 have been the subject of much research in recent years by our laboratories. Streptococcus gordonii has been shown to interact with platelets directly (see Mechanism 1 above). Surface factors Hsa (serine-rich repeat polypeptide) and SspA/SspB (antigen I/II family polypeptides) play critical roles in mediating platelet interactions.56,57,58,59 In a rabbit model of endocarditis, mutation of the hsa gene alone is sufficient to ablate formation of infective vegetations on heart valves (Figure 6).
Figure 6. Heart valve specimens from a catheter-induced rabbit model of IE: (a)S. gordonii wild-type – the white arrow indicates blood clot formation, and the black arrows denote vegetations present on valve leaflets resulting from systemic infection by S. gordonii; (b)S. gordonii hsa: bacteria without the hsa gene appear not to induce IE.
More recently, our studies have identified a new protein (PadA) which binds and activates platelets, causing them to contribute to undesirable functions including blood clotting.60,61,62 Tripeptide motifs, including an NGR (Asparagine-Glycine-Arginine) motif that is also found in human blood protein fibrinogen, are responsible for PadA binding to activated platelets.63 The platelet receptor implicated in PadA binding is integrin αIIbβ3. Together this information suggests that the bacteria are mimicking fibrinogen, effectively fooling platelets into activating and triggering the clotting process inappropriately. It also appears that Hsa and PadA may be reliant on each other for mediating bacterial interactions with platelets and extracellular matrix proteins, such as fibronectin and vitronectin.63 Further recent evidence shows that antibodies targeting Hsa and PadA prevent platelet aggregation and protect against S. gordonii-induced endocarditis in a rat model.64
Proteins with similarities to PadA have been identified in a number of IE pathogens (Streptococcus mitis, Streptococcus oralis, Enterococcus faecalis). It will be important to determine if there is a common functional mechanism that could be targeted to control infection-induced cardiovascular disease. It has already been suggested that PadA and Hsa in S. gordonii could be promising vaccine candidates against viridans group streptococci-induced IE.64
Clinical implications
The appropriate management of dental patients at risk of IE has been a controversial topic over recent years. An increase in IE incidence has occurred after the change in practice regarding antibiotic prophylaxis recommended by NICE in 2008.38,65 In 2015, NICE reviewed the guidelines and announced that there was insufficient evidence to warrant a change, generating considerable debate in the literature.35,66,67,68,69,70,71,72 In addition, there were changes in the law on informed consent in the UK.73 In July 2016, a change in the wording of the NICE guidelines was announced65 meaning that, in individual cases, antibiotic prophylaxis may be appropriate. As discussed elsewhere,74 the current situation still leaves considerable challenges for dentists when faced with implementing the guidelines.
A qualitative study from the USA in 2013 found that dentists were keen to use the associations between oral and systemic diseases as a motivating factor for patients to maintain good oral health.75 In fact, the Editors' Consensus Report of The American Journal of Cardiology and Journal of Periodontology suggests that patients with moderate to severe periodontal disease should be informed about the increased risk of cardiovascular disease.76 In addition, it states that a periodontal assessment should be considered in patients with cardiovascular disease who have tooth loss and unexplained high levels of CRP or other inflammatory markers.76 Certainly, in the case of IE, dentists should reinforce the importance of excellent oral hygiene to patients at risk of developing the condition and encourage regular dental reviews.74 At-risk patients and dentists should all be aware of the symptoms of endocarditis35 and refer early when appropriate.
Conclusion
The importance of maintaining good oral health for systemic health is an ancient concept but just as relevant today. Direct or indirect mechanisms have been proposed for links between oral bacteria and systemic diseases. Recent research has shown direct effects of oral bacteria on platelet function, leading to platelet activation and formation of infected clots. These new discoveries explain how oral microbes stimulate formation of vegetations and this work will lead to the development of new ways to control or prevent the formation of infective blood clots. At-risk patients and dentists should be aware of the recent change in wording to the antibiotic prophylaxis guidelines and be knowledgeable about early IE symptoms. Two and a half millennia after the Ancient Greeks took notice, it is time for the dental and medical professions to raise public awareness about the importance of good oral hygiene to stay healthy.